
Nat Methods:1亿个参数,2万个基因!张学工/马剑竹/宋乐开发单细胞转录组学大规模预训练模型
2024-06-09 测序中国 测序中国 发表于上海

研究团队报道了一个大型的预训练模型scFoundation,其具有1亿个参数,覆盖约20,000个基因,在超过5,000万个人类单细胞转录组谱上进行了预训练。
随着人工智能技术的不断进步,大规模的pre-training(预训练)模型已经在自然语言处理及相关领域的研究带来革命性的变化,并且其应用范围正不断扩展。在生命科学中,生物体有其潜在的“语言”,细胞是人体的基本结构和功能单位,是由DNA、RNA、蛋白质、基因表达值等无数“词语”组成的“句子”。那么是否能够基于大量“句子”开发研究细胞的基础模型呢?
近年来,单细胞转录组测序(scRNA-seq)数据提供了对细胞系统的高通量观察,为建立细胞的基础模型提供了重要参考信息。在转录组学数据中,基因表达谱描述了细胞内基因-基因共表达和相互作用的复杂系统。随着研究的深入,单细胞转录组数据规模呈指数级增长,这与用于训练大型语言模型(LLM)的自然语言文本的数量相当,为训练用于细胞研究的大规模模型提供了基础。
2024年6月6日,来自清华大学张学工、马剑竹团队联合百图生科公司宋乐团队在Nature Methods发表了题为“Large-scale foundation model on single-cell transcriptomics”的文章。在该文章中,研究团队报道了一个大型的预训练模型scFoundation,其具有1亿个参数,覆盖约20,000个基因,在超过5,000万个人类单细胞转录组谱上进行了预训练。分析显示,scFoundation在可训练参数大小、基因维数和训练数据量方面是一个大规模的模型,能够有效地捕获各种细胞类型和状态下基因之间的复杂关系。此外,为了验证scFoundation学习细胞和基因特征的能力,研究人员还进行了多项下游任务的实验。结果表明,scFoundation可以在多种单细胞分析任务中实现最先进的性能,例如基因表达增强、药物反应分类等。因此,该研究揭示了转录组学数据的大规模预训练模型的有效性和价值,并证明了其在促进生物学和医学研究方面的巨大应用价值。该论文第一作者是清华大学自动化系博士生 郝敏升。

文章发表在Nature Methods
主要研究内容
scFoundation预训练模型架构
首先,研究团队开发了xTrimoGene,其具有算法效率和工程加速策略,它包括一个嵌入模块和一个非对称编解码器结构。嵌入模块将连续的基因表达量转化为可学习的高维向量,作为编码器和解码器的输入。该模块完全保留来自原始表达式值的信息,与之前模型中使用的离散值相比有显著改进。非对称编解码器结构是专门为适应单细胞基因表达数据的高稀疏特性而设计的,这一架构对零和非零值给予了不同的关注和计算资源,从而实现了所有基因关系的高效学习,而不需要进行任何选择。
考虑到单细胞基因表达数据在读取深度上存在较大差异,研究团队还设计了一种新的预训练任务,称为读取深度感知(RDA)建模。在RDA中,研究团队训练模型根据其它基因的表达量来预测细胞被掩盖基因的表达。这种预训练过程使预训练模型不仅能够捕获细胞内的基因-基因关系,而且能够协调不同读取深度的细胞。
随后,研究团队收集了所有公开的单细胞数据资源,构建了一个完整的单细胞基因表达数据集,涵盖了不同疾病、肿瘤和正常状态下的100多种组织类型,几乎包含了所有已知的人类细胞类型和细胞状态。经过测试,scFoundation可以在不依赖数据集训练过程的情况下获得最佳性能。
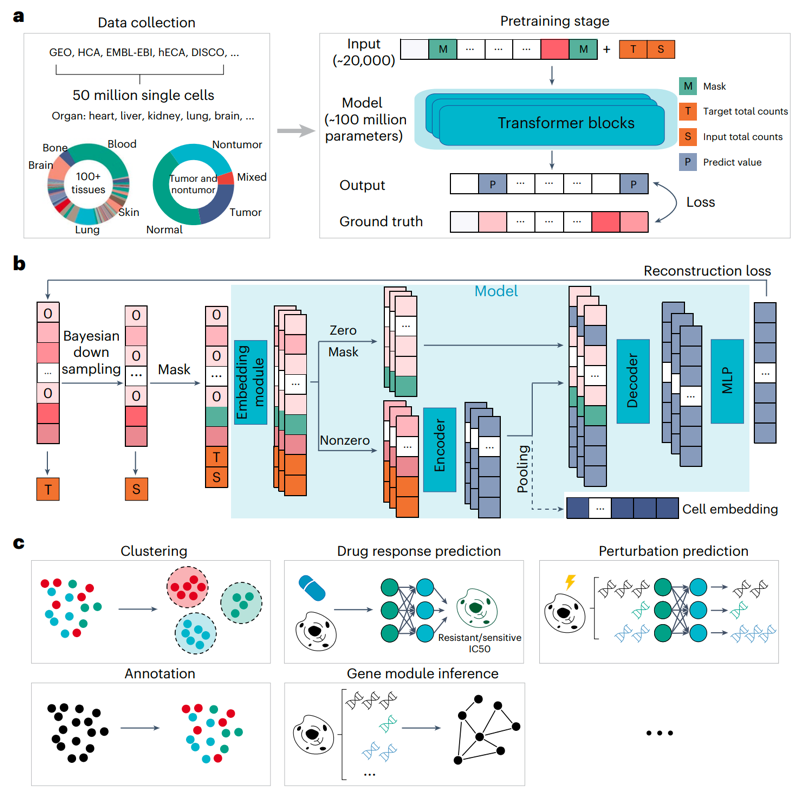
图1. 预训练模型的概括图
scFoundation改善癌症药物反应预测
肿瘤药物反应(CDRs),即研究肿瘤细胞对药物干预的反应。预测CDR对于指导抗癌药物设计和理解癌症生物学至关重要。因此,研究团队将scFoundation与CDR预测方法DeepCDR结合,在多个细胞系数据中预测药物的半最大抑制浓度IC50值,使用scFoundation提取转录组特征并将其输入到后续的预测模块中。
结果发现,大多数药物和所有癌症类型通过使用scFoundation的嵌入实现了更高的皮尔森相关系数(PCC)。研究团队还进一步可视化了药物和癌症类型的最佳预测案例,表明无论lC50高或低,基于scFoundation嵌入的DeepCDR模型都可以预测准确,并实现了0.93以上的PCC。
接下来,研究团队进一步将药物分为不同的治疗类型,以检验IC50预测性能是否与它们的内在机制有关。研究团队观察到,根据scFoundation预测的结果,化疗药物(如抗肿瘤抗生素和拓扑异构酶抑制剂)比靶向治疗药物(如ATM和PARP抑制剂)具有更高的PCC,这可能是由于特定的基因突变往往对靶向治疗有重要影响,但突变信息很难从基因表达数据中被揭示。总之,以上发现说明了scFoundation在扩展对癌症生物学中药物反应方面的潜力,并可指导设计更有效的抗癌治疗。此外,scFoundation还可将药物敏感性预测模型转移到单细胞数据中,有效地促进了药物基因组学信息从细胞系到单细胞数据的转移。
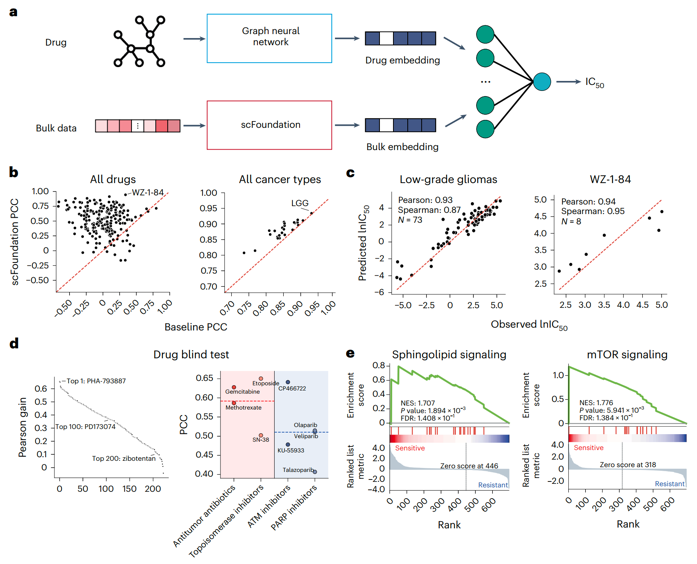
图2. 基于scFoundation嵌入的药物反应预测
scFoundation预测更准确的扰动反应
细胞对扰动的反应对于生物医学应用和药物设计至关重要,因为其有助于识别不同细胞类型和潜在药物靶点之间的相互作用。研究团队将scFoundation与一个先进的模型GEARS结合起来进行扰动预测任务。在原始的GEARS模型中,基因共表达图与扰动信息相结合,预测扰动后的基因表达,共表达图中的每个节点代表一个基因,初始嵌入是随机的,边缘连接共表达基因。研究人员从scFoundation解码器中获得每个细胞的基因上下游嵌入信息,并将其设置为图中的节点。
随后,研究团队在三个扰动数据集上训练和测试了模型,并计算了前20个差异表达基因与扰动后基因表达谱之间的均方误差,以评估模型的性能。与原始的GEARS基线模型相比,该联合模型在所有基因扰动数据集上获得了更低的均方误差值。此外,研究团队进一步分析了预测值落在基因真实平均表达值百分之五以内的比例,发现基于scFoundation模型的百分比更高,说明其提供了更合理的扰动后基因表达值分布,对基因扰动的分析还强调了该模型准确分类不同类型遗传相互作用的能力。
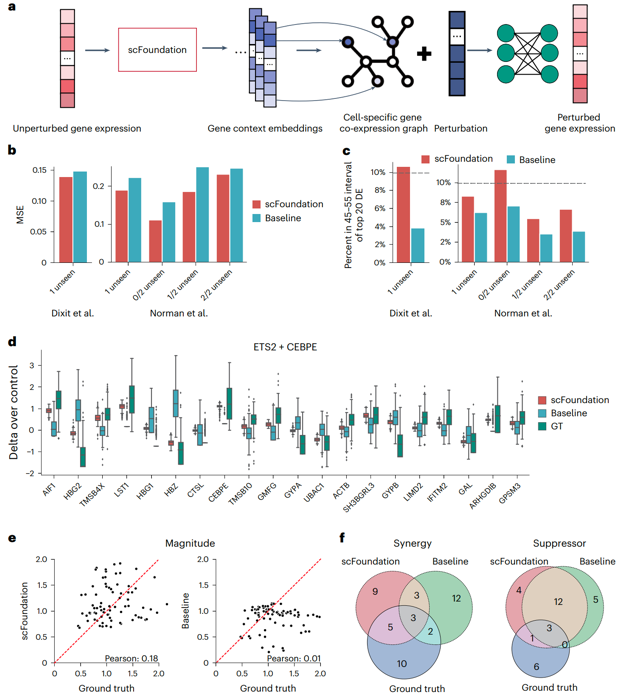
图3. 基于scFoundation嵌入的基因扰动预测
结语
该研究报道了最新开发的大规模预训练模型—scFoundation,该模型具有惊人的1亿参数规模,并经过对超过5000万个人类单细胞转录组数据的训练。scFoundation不仅具备前所未有的规模和基因维度,还为单细胞转录组学提供了丰富的分子特征观测数据。基于多个层面的应用,scFoundation在多种生物医学任务中均具有卓越性能,包括组织药物反应预测、扰动反应预测等。因此,未来scFoundation不仅可以协助研究人员深入研究不同细胞类型及其在不同条件下的基因表达模式,还可能在众多生物医学任务中取得显著的突破。
论文原文
Hao M, Gong J, Zeng X, Liu C, Guo Y, Cheng X, Wang T, Ma J, Zhang X, Song L. Large-scale foundation model on single-cell transcriptomics. Nat Methods. 2024 Jun 6. doi: 10.1038/s41592-024-02305-7.
https://doi.org/10.1038/s41592-024-02305-7
 分享
分享
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#单细胞转录组# #训练模型#
69 举报