
【AJH】蕈样肉芽肿、Sézary综合征、皮肤B细胞淋巴瘤的诊断、风险分层和治疗(2025更新)
2025-07-19 聊聊血液 聊聊血液 发表于上海

近日《American Journal of Hematology》发表综述,阐述了蕈样肉芽肿、Sézary综合征、皮肤B细胞淋巴瘤的诊断、风险分层和治疗,现翻译整理供参考,共约1.9万字。
原发性皮肤淋巴瘤指一组异质性T细胞/B细胞淋巴瘤,其中蕈样肉芽肿(mycosis fungoides,MF)是最常见类型;而Sezary综合征(Sezary syndrome,SS)是皮肤T细胞淋巴瘤的一个变种,以红皮病及循环性肿瘤性T细胞相关白血病为主要特征。
近日《American Journal of Hematology》发表综述,阐述了蕈样肉芽肿、Sézary综合征、皮肤B细胞淋巴瘤的诊断、风险分层和治疗,现翻译整理供参考,共约1.9万字。

疾病概述
原发性皮肤淋巴瘤是一组异质性的结外非霍奇金淋巴瘤,在世界卫生组织(WHO)和国际共识分类(ICC)的最新分类中进行了分类(表1)。

大多数(约70%)原发性皮肤淋巴瘤起源于T细胞,包括MF和SS。相比之下,B细胞来源的皮肤淋巴瘤可分为三大类:原发性皮肤滤泡中心淋巴瘤(PCFCL)、原发性皮肤弥漫大B细胞淋巴瘤,腿型(PCDLBCL,LT)以及原发性皮肤边缘区淋巴瘤/淋巴增殖性疾病(PCMZL/LPD)。EB病毒阳性黏膜皮肤溃疡(EBVMCU)是一种最近定义的EB病毒阳性淋巴增殖性疾病,影响皮肤和黏膜。原发性皮肤T细胞淋巴瘤(CTCL)和原发性皮肤B细胞淋巴瘤(CBCL)的发病率一直在增加,但目前每百万人中仍不到10例,发病率最高的群体是男性和50岁以上成年人。
遗传学证据表明紫外线辐射是CTCL的风险因素,流行病学研究表明,接触环境毒素也可能会增加患病风险。某些特定的药物以及可能的COVID-19疫苗接种也与CTCL有关。抗原或细胞因子驱动的T细胞淋巴增殖或异常或可解释这些罕见的、可能是医源性的CTCL病例。
少数家族性MF的报告以及在散发性和家族性MF中检测到特定的II类HLA等位基因表明,宿主遗传因素可能在MF的发病中起作用。CTCL患者继发恶性肿瘤的发病率更高,包括黑色素瘤,因此需要进行适当的筛查和监测。CBCL患者也更容易发生后续肿瘤,包括女性的甲状腺癌、肾癌和肺癌,以及男性的黑色素瘤、膀胱癌和前列腺癌。
通过下一代测序(NGS),我们对CBCL和CTCL的遗传和分子发病机制有了更多了解。涉及淋巴细胞特异性(包括抗原和细胞因子受体依赖性)信号通路、表观遗传和染色质重塑蛋白以及细胞周期调节因子的改变反复出现,并具有治疗意义。皮肤屏障的破坏和皮肤微生物组的变化在CTCL中具有临床和治疗价值,而局部(在淋巴瘤微环境中)和全身(“宏观环境”)免疫失调在CBCL和CTCL的发病中都起着病理作用。
诊断
CBCL
诊断和分类CBCL需要进行切开活检、切除活检或4–6毫米穿孔活检,包括网状真皮和皮下脂肪,用于形态学和免疫组化分析,并进行分期评估以排除系统性疾病。使用适当的免疫组化染色(例如CD5、细胞周期蛋白D1)也有助于区分CBCL和系统性淋巴瘤继发皮肤受累。
PCFCL
PCFCL通常表现为单个紫色至粉红色的丘疹或结节(通常活检以排除非黑色素瘤皮肤癌),或表现为多发性丘疹、斑块或肿瘤,边缘有红晕,多发生在中年成年人的前额、颈部和上背部。虽然可以看到聚集性病变,但多灶性疾病较为少见。其他不太常见的表现形式包括酒渣鼻和瘢痕性脱发,伴有肿胀性粉红色斑块。皮肤镜有助于区分这些皮肤淋巴瘤与非黑色素瘤皮肤癌。组织病理学上,PCFCL的特征为滤泡性、弥漫性或混合性生长模式,由大中心细胞和可变的中心母细胞组成,这些细胞源自生发中心B细胞。与系统性滤泡性淋巴瘤不同,大多数PCFCL不携带涉及bcl-2位点的t(14;18)易位,并且在免疫组化中不强表达bcl-2,但也有少数病例可能表达。bcl-2和CD10的强表达可能提示滤泡性淋巴瘤的继发性皮肤受累,而bcl-2在超过75%的细胞中表达可能与PCFCL的皮肤复发风险增加有关。PCFCL表达bcl-6,可变表达CD10,且为MUM-1/IRF-4、FOXP1、IgM阴性,与其源自生发中心B细胞的起源相一致。
学者对PCFCL(n=30)和继发性皮肤受累的系统性滤泡性淋巴瘤(n=10)进行了全外显子测序。与之前的观察一致,大多数(即73%)PCFCL不表达bcl-2,与继发性皮肤滤泡性淋巴瘤相比,其增殖性更强,可通过Ki67表达来确定。值得注意的是,继发性皮肤滤泡性淋巴瘤中表观遗传修饰因子CREBBP、KMT2D、EZH2或EP300的复发性突变较为常见,但在PCFCL中很少观察到。因此,在63%的继发性皮肤滤泡性淋巴瘤病例中观察到至少两个基因的突变,但在PCFCL中很少见。相比之下,TNFRSF14在PCFCL中频繁突变。
最近一项使用低覆盖度全基因组测序的研究发现,系统性滤泡性淋巴瘤更有可能出现18号染色体的扩增,尤其是在BCL2位点。此外,PCFCL的远处扩散与基因组不稳定性、REL和XPO1的扩增以及CDKN2A和CDKN2B的缺失有关。因此,遗传学特征有助于区分PCFCL和继发性皮肤滤泡性淋巴瘤。
最近描述了一种涉及女性下生殖道(宫颈和/或阴道)的新变体,其免疫表型(例如,>80%的Bcl-2阴性)、遗传学(例如,未发现CREBBP、KMT2D突变)和临床特征(局限性疾病,5年总生存率为100%)与PCFCL相似。值得注意的是,一些具有弥漫性生长模式和大中心细胞的PCFCL病例很难与弥漫大B细胞淋巴瘤(DLBCL)区分。这些病例可能具有高增殖指数。然而,滤泡性生长区域、与中心细胞相符的裂解细胞、使用免疫组化(例如CD21、CD23、CD35、SSTR2A、D-240)的滤泡树突细胞网络、大量T细胞以及缺乏BCL2、MUM1、FOXP1、IgM和C-MYC的表达,这些都倾向于支持PCFCL的诊断。
PCDLBCL,LT
PCDLBCL,LT通常影响老年女性,表现为迅速进展的红棕色至蓝色肿瘤,累及一条或两条下肢。肿瘤可能形成溃疡,较大的肿瘤周围可能有较小的卫星病变;相对罕见的表现形式包括疣状和多色结节。基于人群的研究表明,仅限于腿部的受累比“腿型”这一名称所暗示的要更少见,因为其他皮肤部位的受累也相当常见。这些淋巴瘤的特征为弥漫性片状的中心母细胞和免疫母细胞,这些细胞不累及表皮,但经常深入真皮和皮下组织。与PCFCL不同,淋巴瘤细胞高度表达bcl-2,可能是由于基因扩增所致,因为t(14;18)在PCDLBCL,LT中未被观察到。Bcl-2过表达或bcl-2和c-myc的双重表达与较差的总生存率相关。c-myc和bcl-2易位(“双打击”)在PCDLBCL,LT中比例不到20%。大多数病例为MUM-1/IRF-4、FOXP1、IgM和bcl-6阳性,CD10阴性,基因表达谱类似于活化B细胞。EB病毒原位杂交(EBER)为阴性。也许并不令人惊讶的是,PCDLBCL,LT的遗传学特征与活化B细胞型弥漫大B细胞淋巴瘤(ABC-DLBCL)相似,在CD79B、CARD11和MYD88中观察到NF-κB激活突变。其中,体细胞MYD88 L265P突变最为常见,发生率约为75%,并且在PCFCL中很少或从未观察到。约30%的病例中观察到MYC重排,与较差的预后相关。尽管使用体细胞高突变的免疫球蛋白重链可变区(IGHV),但IGHV内的金黄色葡萄球菌超抗原结合位点得以保留,从而暗示了超抗原依赖的B细胞受体信号传导在疾病发病机制中的作用。
PCMZL/LPD
PCMZL/LPD患者通常表现为躯干、手臂和头颈部的单个无症状粉红色丘疹,或表现为累及躯干和手臂的红棕色多灶性丘疹、斑块或结节。尽管在欧洲观察到与伯氏疏螺旋体(Borrelia burgdorferi)的关联,但在美国的病例中并未发现类似相关性。PCMZL/LPD由小的、“中心细胞样”边缘区B细胞、单核细胞样B细胞、淋巴浆细胞、浆细胞、类似中心母细胞和免疫母细胞的细胞、反应性T细胞、嗜酸性粒细胞和组织细胞组成的混合性浸润,成分比例可变。边缘区B细胞特征性地表达bcl-2,可能表达CD43,但缺乏bcl-6、CD10、CD5和CD23的表达。最近将PCMZL/LPD分为两个亚组,其中大多数PCMZL/LPD表达类别转换的免疫球蛋白,并且病程较为惰性。最近的分类系统建议将这种类型的PCMZL视为淋巴增殖性疾病(LPD),以反映其与非典型反应性B细胞浸润(假B细胞淋巴瘤)在临床和组织病理学上的重叠,以及极其良好的预后。该组的特征是B细胞聚集,周围有浆细胞,大量混合的T细胞和反应性滤泡,可能代表对某种抗原刺激的反应。第二组表达IgM,行为类似于结外边缘区淋巴瘤,常有复发和结外扩散。这一组通常表现为弥漫性浸润,浆细胞混杂其中而非周围,滤泡频繁受累,更有可能累及皮下组织,并且更有可能转化为弥漫大B细胞淋巴瘤。PCMZL/LPD常携带FAS、SLAMF1、SPEN和NCOR突变以及涉及FOXP1和BCL10的IGH易位。部分PCMZL/LPD在临床和组织病理学上与原发性皮肤CD4阳性小或中等淋巴增殖性疾病有重叠特征,这些病例以小到中等大小的淋巴样细胞为主,可能表现出增加的PD-1和CD4阳性T细胞,以及IGH和TRG的克隆性重排。PCMZL复发的可能性更大,此外在原发性皮肤CD4阳性小/中等淋巴增殖性疾病中未发现TNFAIP3和FAS突变。
EBVMCU
EBVMCU通常表现为皮肤或黏膜上的单个、疼痛且边界清晰的溃疡,黏膜包括口咽部、生殖器黏膜或胃肠道。患者通常处于免疫抑制状态,原因可能是与年龄相关的免疫衰老、医源性免疫抑制(尤其是使用甲氨蝶呤)、实体器官移植、HIV感染或原发性免疫缺陷。通常没有其他症状,也没有骨髓受累、淋巴结肿大或肝脾肿大。此外,外周血中EB病毒并不增加。病变可能与先前的创伤有关,创伤相关的炎症可能有助于发病机制。组织损伤和坏死伴纤维素样改变可能导致免疫隔离区域和局部EB病毒增殖。
组织病理学上,EBVMCU表现为浅溃疡,其下有混合性浸润,包括大量浆细胞、组织细胞和嗜酸性粒细胞,底部由大量CD8+ T细胞组成。在其中散布着大小不一的B细胞免疫母细胞,其中部分具有霍奇金-里德-斯特恩伯格样(HRS样)外观;也可能存在凋亡的浆细胞样细胞。免疫组化表明,免疫母细胞是非生发中心B细胞,表达CD20、PAX5、OCT2和MUM-1,CD10阴性。它们CD30阳性,可能表达CD15。浆细胞可能有轻链限制。EB病毒原位杂交(EBER)标记免疫母细胞,包括HRS样细胞以及其他较小的淋巴样细胞。血管侵犯和坏死通常存在。B细胞受体基因重排研究显示,在不到半数病例中存在克隆性B细胞群体;T细胞受体基因重排研究可能表现为寡克隆性或克隆性。
EBVMCU的组织病理学鉴别诊断包括EBV+弥漫大B细胞淋巴瘤和霍奇金淋巴瘤,以及多形性淋巴增殖性疾病;临床特征对于正确诊断至关重要,尤其是在小活检标本中。最近的一项NGS研究表明,EBVMCU像EBV+弥漫大B细胞淋巴瘤一样,表现出广泛的基因变异。与克隆性造血相关的TET2突变较为常见。
CTCL
MF
MF的明确诊断(特别是斑块/斑疹期)具有一定挑战性,因其许多临床和病理特征为非特异性的,并且与反应性过程重叠。许多患者在获得明确诊断之前,其症状可能归因于湿疹、银屑病或副银屑病多年。在回顾性系列中,从症状出现到诊断的中位时间长达3-4年,也可能超过四十年。在这段时间里,患者可能被误诊为其他疾病,特别是特应性皮炎和银屑病。鉴于MF的诊断需要临床和病理学的关联,以及特定组织病理学发现与诊断的可变关联,活检报告不时地“提示”诊断。这种活检报告中偶尔的不确定性以及缺乏更明确的组织病理学诊断,可能让不熟悉MF诊断挑战性的临床医生感到沮丧。此外,在活检时使用皮肤定向治疗,包括局部皮质类固醇,可能减少或消除肿瘤T细胞和其他组织病理学发现,进一步增加诊断的难度,因为这些治疗可在2-4周内减少或消除肿瘤T细胞和关键的组织病理学发现。药物反应、慢性海绵状皮炎、结缔组织疾病、硬化性苔藓和色素性紫癜性皮病只是少数可能模仿MF的疾病。虽然MF的明确诊断可以仅根据临床和组织病理学特征来做出,但通过免疫组化确定T细胞克隆性和评估T细胞抗原(例如CD2、CD5、CD7)表达的异常丢失也有用。基于PCR的方法能够检测甲醛固定、石蜡包埋活检标本中T细胞受体(TCR)的克隆性重排。虽然基于PCR的方法较为敏感,但应该谨慎解释,因为克隆性TCR基因重排可能在正常老年人和患有良性皮病或其他疾病状态的患者中被检测到。然而,从两个不同部位检测到相同的克隆对于MF来说是相当特异的。然而,即使这个特征也存在问题的,因为罕见的反应性过程在多次活检中显示,通过基于PCR的基因重排似乎是相同的T细胞克隆。此外,部分MF病例可能没有可检测的T细胞克隆。最近的研究表明,NGS可能对评估MF/SS中的T细胞克隆性更敏感和/或更特异,但尚未广泛可用。此外,NGS可能与基于PCR的研究有类似的陷阱,因为它可能在反应性浸润中识别克隆性T细胞,并且可能无法在CTCL中识别克隆性T细胞。MF/SS在多大程度上可能被类似于单克隆B细胞淋巴细胞增多症(MBL)或意义未定的单克隆丙种球蛋白病(MGUS)的癌前状态所先兆,这个问题仍有争议,并且定义并不明确。
MF/SS中的恶性淋巴细胞通常为CD3+CD4+和CD8-,但经常失去其他泛T细胞抗原的表达。因此,显示大量CD4+细胞缺乏CD2、CD5和/或CD7表达对MF具有高度特异性(特异性>90%)。然而,反应性皮病也可能显示CD4阳性T细胞的主导地位和CD7的丢失或低表达,MF多丢失T细胞抗原,这些结果必须谨慎解释。发现CD4阳性T细胞的显著优势,特别是通过表皮趋向性T细胞,有助于支持MF的诊断。同样,发现广泛的CD7丢失、表皮T细胞中泛T细胞抗原的优先丢失或多个泛T细胞标记的丢失,在具有挑战性的病例中也可支持MF的诊断。在临床上,斑块/斑疹阶段的MF通常以持续性和进行性病变为特征,这些病变在“泳衣”分布中发展,大小、形状和颜色各不相同。这些病变通常为大的(>5厘米)、瘙痒的和多灶性的“经典型”MF。在有色皮肤(SoC)中,病变呈多色,包括超色素和低色素的斑块/斑疹。Espinosa等人确定超色素沉着、苔藓化和银色光泽在SoC中显著更常见。然而,也报道了广泛的MF变体,其差异在于趋向性(例如,毛囊MF,腺趋向性),分布(例如,掌跖MF),外观(例如,松弛皮肤,色素性皮病),色素沉着(例如,低色素和超色素变体)和局灶性(例如,单发性MF)。组织病理学上,斑块/斑疹MF的特征为增大、表皮趋向性淋巴细胞,其核不规则,通常在真皮中呈带状分布,与密集的胶原条带(“电线”纤维化)相关。在表皮中,称为Pautrier微脓肿的肿瘤T细胞聚集体在少数病例中被看到,但对诊断有用。毛囊趋向性和/或汗腺趋向性可能在少数病例中被看到。最近的研究表明,包括临床信息(包括照片)可提高病理学家的诊断准确性,从而突出了临床信息对于准确的组织病理学诊断的重要性。
SS
传统上, SS被定义为一种与红皮病、难以控制的瘙痒、外翻性睑外翻以及掌跖角化症相关的白血病型CTCL。20世纪早期至中期的一系列研究,始于Sézary 1938年的最初标志性观察,识别出在MF或SS患者外周血中存在一种具有沟槽状、分叶(即“脑回状”)核的大型淋巴细胞群体。与其他慢性淋巴增殖性疾病一样,Sézary细胞计数最好以绝对值表示,目前在ISCL/USCLC/EORTC TNMB分期分类中,≥1000个细胞/μL被归类为B2期疾病,但这种仅依赖绝对值来定义疾病负荷的方式可能会错误分类(低估)少数患者。在外周血中检测到Sézary细胞的形态学特征并不特异于CTCL,因为正常供体的外周血以及良性疾病中也可能发现Sézary细胞。
皮肤的组织病理学表现通常与MF中观察到的相似,表皮趋向性不那么显著,尽管皮肤活检的结果可能出乎意料地细微且不特异。与MF一样,免疫组化研究显示CD4优势以及泛T细胞标记物的丢失可能有助于诊断。淋巴结受累的特征为淋巴结结构被浸润的Sézary细胞完全破坏。
在SS中,克隆性T细胞通常通过多色流式细胞术表现为CD3+CD4+和CD8−。与MF一样,常可观察到泛T细胞抗原(包括CD2、CD3、CD4、CD5、CD7和/或CD26)的异常丢失。其中CD7和/或CD26表达的异常丢失最为常见,出现在大多数病例中。外周血中超过40%的CD4+ T细胞丢失CD7和CD26表达对SS具有100%的特异性。分子学检测,包括通过PCR检测克隆性T细胞受体(TCR)基因重排以及克隆性细胞遗传学异常的存在,为T细胞克隆性可提供证据。另一种用于证明T细胞克隆性的方法是采用针对不同TCR β链可变区家族成员(TCR-Vβ)的抗体组合进行多色流式细胞术,如果克隆性T细胞群体显著高于携带相同Vβ链的多克隆T细胞的背景频率,则可以成功识别克隆性T细胞群体。β链恒定区包含两个基因片段(C1和C2)。类似于B细胞淋巴增殖性疾病中的κ或λ轻链限制,在αβ T细胞克隆性中,β链恒定区-1(TRBC1)的过度(或不足)表达是一种敏感且特异的生物标志物。通过FISH检测CTCL中反复扩增或缺失的驱动基因可有信息价值,但尚未广泛使用。
除了ISCL/USCLC/EORTC标准外,最新的WHO分类还要求出现红皮病、全身性淋巴结肿大以及皮肤、外周血和淋巴结中克隆相关的T细胞(Sézary细胞)。在极少数情况下,SS可能发生在之前有MF斑块/斑疹病史的患者中。ISCL建议将此类病例指定为“MF后继发的SS”或“继发性红皮病性CTCL”。相反,没有红皮病的MF患者可能符合SS的血液学标准。在这种情况下,建议使用“MF伴白血病受累”这一名称。
非MF/SS的CTCL亚型
患者初始诊断期间的一个重要目标是区分非 MF/SS CTCL 亚型和MF/SS,因为每个非 MF/SS 淋巴瘤的自然史、预后和治疗方法具有高度变异性。这些 CTCL 亚型的详细描述超之前已有文章总结了这些 CTCL 亚型的的显著特征(R. Willemze, L. Cerroni, W. Kempf, et al., “The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas,” Blood 133 (2019): 1703–1714; R. A. Wilcox, “Cutaneous T-Cell Lymphoma: 2011 Update on Diagnosis, Risk-Stratification, and Management,” American Journal of Hematology 86 (2011): 928–948; R. Dummer, M. H. Vermeer, J. J. Scarisbrick, et al., “Cutaneous T Cell Lymphoma,” Nature Reviews. Disease Primers 7 (2021): 61.)。
风险分层
CBCL
分期建议包括病史采集、体格检查和影像学检查(CT、PET或PET/CT)。对于PCDLBCL,LT患者,以及有其他CBCL且伴有不明原因的血细胞减少症的患者,应进行骨髓活检和穿刺。虽然TNM分期分类可描述疾病的范围,但在CBCL中,分期对预后的价值有限,因为疾病组织病理学是风险分层的主要决定因素。这一点在一项基于人群的研究中得到了强调,该研究发现组织病理学和皮肤受累部位是重要的预后因素。相比之下,国际结外淋巴瘤研究小组在PCFCL和PCMZL患者中识别出三个独立的预后因素(即LDH升高、>2个皮肤病变和结节性病变),这些因素被合并形成了皮肤淋巴瘤国际预后指数(CLIPI)。没有任何不良预后因素的患者,其5年PFS为91%,而存在两个或三个不良预后因素的患者为48%。由于大多数复发局限于皮肤,CLIPI无法通过OS对患者进行风险分层。在欧洲系列研究中,多发性皮肤病变与较差的无病生存率相关,但在北美大型系列研究中,多发性皮肤病变并未与无病生存率相关。在皮肤B细胞淋巴瘤中,风险分层的最重要因素仍然是组织病理学分类。惰性CBCL(PCFCL和PCMZL)与5年疾病特异性生存率≥95%相关,尽管表达IgM的PCMZL与较不理想的病程相关。在PCMZL/LPD中,皮肤和血液中存在匹配的B细胞克隆,或存在副蛋白血症,与皮肤复发或预后无关。在PCFCL中,生长模式的差异、中心母细胞的密度以及细胞遗传学发现似乎并未提供有意义的预后信息。尽管罕见,但部分系统性FL可能最初表现为皮肤受累,但其结外病变低于影像学检测阈值。CREBBP、KMT2D、EZH2、EP300(如系统性FL中所见)的突变、涉及bcl-2基因的重排以及低增殖率(Ki67<30%)可能有助于识别这些患者。相比之下,PCDLBCL,LT的5年疾病特异性生存率约为50%,bcl-2和c-myc的双表达以及CDKN2A的丢失与较差的生存率相关。与仅表现为单个肿瘤的患者相比,累及一个或两个腿部的多部位受累与显著较差的疾病特异性生存率相关。
虽然EBVMCU可能会局部扩散或复发,但远处播散极为罕见,如果观察到,应考虑其他EBV相关淋巴增殖性肿瘤的可能性。值得庆幸的是,EBVMCU通常为局部病变,预后良好,因为大多数病例在减少免疫抑制后会自行消退。
CTCL
与许多其他淋巴增殖性疾病(其中细胞遗传学和实验室检查结果在风险分层中起重要作用)不同,TNMB(肿瘤、淋巴结、转移、血液)分期在MF/SS中仍然是重要的预后因素,并构成了“风险适应性(risk-adapted)”治疗方案的基础。仅表现为斑块/斑疹的患者为I期疾病,但可根据皮肤受累程度进一步分为IA期(<10%体表面积受累或T1)或IB期(>10%体表面积受累或T2),并根据病变是斑块期(T1a/T2a)还是斑疹期(T1b/T2b)进行分类。从实用角度出发,患者一只手的面积(包括手掌和手指)大约代表1%的体表面积。目前的分期和诊断建议不要求对临床上正常的淋巴结进行活检;但建议对任何异常淋巴结(直径≥1.5厘米或质地坚硬/固定)进行切除活检或核心针活检,优先选择引流受累皮肤区域的最大淋巴结,或在FDG-PET成像中标准化摄取值(SUV)最高的淋巴结。尽管对于T1或T2期疾病且体格检查未发现淋巴结肿大的患者,淋巴结的影像学检查可作为可选项,但最近的一项国际研究发现,体格检查可能会遗漏影像学上增大的淋巴结,从而导致少数患者的分期发生显著变化,尤其是有斑块的患者。对于斑块/斑疹期疾病(T1/T2)且任何临床上异常淋巴结结构保持完整的患者,应归类为IIA期。总体而言,I-IIA期患者为“局限性(或早期)”疾病,因为这些患者的OS以数十年为单位,I期患者的生存率与同龄正常对照组相似。在诊断时,大多数MF患者为局限性疾病,而患有肿瘤期疾病(T3)、红皮病(T4)、淋巴结受累表现为部分或完全结构破坏(N3)、内脏转移(M1)或显著白血病受累(B2)的患者为“进展期(晚期)”。虽然欧洲医学肿瘤学会(ESMO)和EORTC建议对所有MF/SS患者进行外周血流式细胞术检查,但最近的共识建议支持在外周血流式细胞术在特定患者中使用:患有进展期(≥IIB)疾病、难以控制的瘙痒、全身性斑块/斑疹、红皮病、淋巴细胞增多、LDH升高或对皮肤定向治疗无反应的患者。不幸的是,这些患有更广泛疾病的患者的中位生存期约为1-5年。大细胞转化(LCT)定义为异常T细胞的受累程度超过25%,且其大小至少是正常淋巴细胞的4倍,其基因组复杂,转录上具有独特性,且与不良预后相关。
一项纳入1398例MF患者(其中71%为斑块/斑疹期疾病)和104例SS患者的研究验证了分期分类的价值,因为TNMB分期与OS和疾病特异性生存率显著相关。最近的一项荟萃分析报告了类似的5年OS趋势。尽管最近批准的药物对OS的影响尚不确定,但在接受这些药物治疗的部分患者中观察到的持久反应可能为治疗带来乐观的理由。对于早期患者,男性性别、年龄>60岁、斑块期或毛囊趋向性疾病以及淋巴结N1/Nx期为不良预后因素,并生成早期疾病患者的皮肤淋巴瘤国际预后指数(CLIPi);低危(0-1个危险因素)患者的10年OS率为90.3%,而高危(3-5个危险因素)患者的10年OS率为48.9%。同样,男性性别、年龄>60岁、B1/B2期或N2/N3期疾病以及内脏受累为进展期患者的不良预后因素;低危患者的10年OS率为53.2%,而高危患者的10年OS率为15.0%。
在一项大型国际系列研究(n = 1275)中,IV期疾病、年龄>60岁、LCT和LDH升高确定为进展期MF/SS的独立不良预后因素,并合并到一个预后指数中。低危(0-1个危险因素)患者的5年OS率(68%)优于高危(3-4个危险因素)患者(28%)。对于毛囊趋向性MF患者,提出了另一种分期系统,识别出一组皮肤受累有限且预后更佳的患者。鉴于TNMB分期在风险分层和定义疾病负荷中的重要性,建议使用TNMB分期来定义疾病的初始、最大和当前负荷,其最终将在选择皮肤定向或全身治疗方案中发挥重要作用。在未来,预计对遗传学景观的更好理解将进一步改善风险分层,并导致在CTCL中选择治疗。
治疗
CBCL
由于缺乏随机对照试验,CBCL的治疗建议主要基于小型回顾性研究和机构经验。EORTC和ISCL已发布与NCCN指南一致的共识治疗建议。在大多数情况下,理想的患者管理需要多学科协作,包括皮肤科、内科肿瘤学和放射肿瘤学。
PCFCL
对于患有单个病变的患者,低剂量放疗安全且极为有效,完全缓解率接近100%。对于可以纳入多个放射野的多发性病变患者,放射治疗似乎并不逊色于多药化疗。在一项大型北美系列研究中,仅用放射治疗对惰性CBCL的局部控制率为98%。在同一研究中,仅接受手术切除的患者中有25%观察到需要放疗的局部复发。将放疗推迟到疾病复发时似乎并未影响疾病特异性生存率或OS。因此,单独进行完全切除,并将放射治疗推迟到疾病复发时,也是合理的。对于病变内治疗(例如,皮质类固醇或针对DLBCL的利妥昔单抗),建议“观察等待”的患者需要密切的临床随访。对于皮肤受累范围更广泛的患者,单药利妥昔单抗可有效管理。大约三分之一的患者在接受放疗或单药利妥昔单抗治疗后可能会复发,但复发通常局限于皮肤,并且处理方式与PCFCL的初始管理类似。
PCMZL/LPD
PCMZL患者的管理与PCFCL的初始管理类似。对于单个或少数病变的患者,放疗或手术切除的反应率很高。然而有研究发现,与接受放疗的患者相比,接受手术切除的患者复发率有所增加。对于皮肤受累更广泛的患者,可以选择观察。一旦出现症状,可对病变进行放疗或手术切除。与PCFCL一样,对于有症状且皮肤受累广泛的患者,可使用单药利妥昔单抗。对于伯氏疏螺旋体相关PCMZL患者,建议进行抗生素的初步试验,但对于北美患者价值较小。
PCDLBCL,LT
如前所述,PCDLBCL,LT的自然病程更接近于系统性DLBCL,因此这些患者使用R-CHOP(联合或不联合放疗)。尽管文献中关于这方面的报道较少,但R-CHOP与高危系统性DLBCL患者报告的无病生存率相当。许多患者在单个部位有局限性病变,并像局限性系统性DLBCL患者一样,接受R-CHOP和受累野放疗。复发性疾病的管理与复发性系统性ABC-DLBCL相似(例如,来那度胺、伊布替尼或坦昔妥单抗/来那度胺)。在一项小型II期研究(n=19)中,复发/难治性PCDLBCL,LT患者接受单药来那度胺治疗的6个月总反应率为26%,但在没有MYD88L265P突变的患者中显著更高。免疫检查点(PD-1)阻断的疗效最近有报道,因为在5例复发/难治性患者中,有3例在接受PD-1阻断后获得持久反应。
EBVMCU
在免疫抑制相关EBVMCU中,绝大多数在停用或减少免疫抑制后会自发消退。对于与年龄相关的免疫衰老相关的病例,以及在减少免疫抑制后出现复发和缓解过程的病例,局部放射治疗或单药利妥昔单抗均极为有效。
CTCL
局限期MF的治疗
由于大多数CTCL患者表现为斑块/斑疹期MF,并且预后良好,治疗的最初目标是改善症状和生活质量,同时避免治疗相关的毒性。对于许多患者来说,这可能涉及预期管理(即“观察等待”)或皮肤定向治疗(图1)。
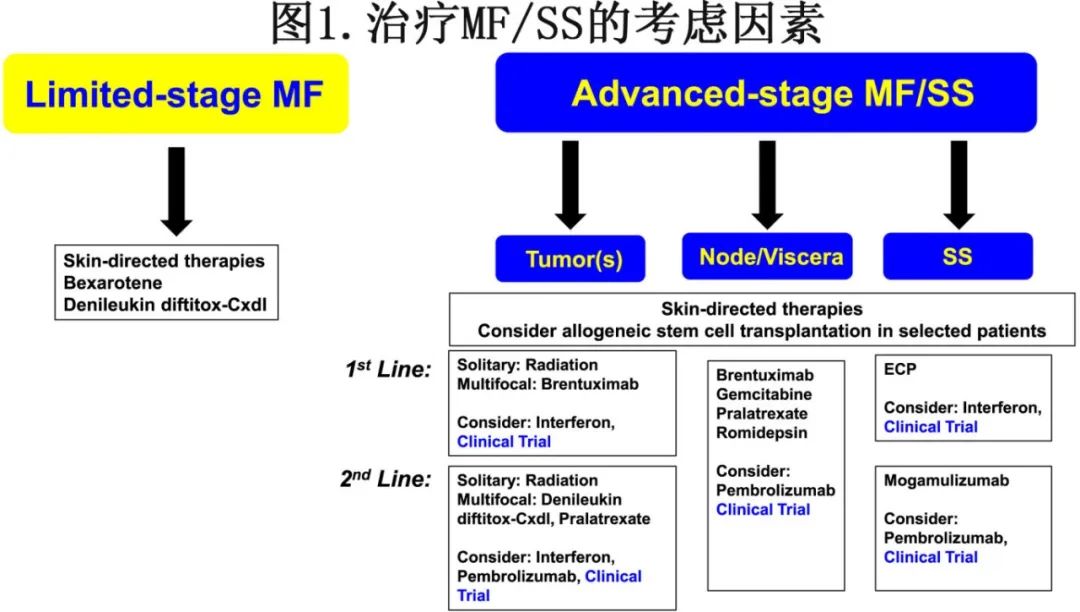
在这些患者中,系统治疗并不能提高反应率或生存率。一项国际前瞻性研究比较了皮肤定向治疗(局部皮质类固醇、紫外线B、补骨脂素加紫外线A、局部氮芥、局部卡莫司汀和局部放射治疗)与系统治疗(口服视黄酸类药物、口服贝沙罗汀、甲氨蝶呤、干扰素和体外光化学疗法)在早期MF中的效果。接受皮肤定向治疗的患者总反应率更高。化疗的有限疗效在回顾性研究也得到强调,这些研究中单药或联合化疗后至下次治疗的中位时间≤4个月。因此,需要治疗的局限期患者最好采用皮肤定向治疗,通常在皮肤科医生和/或放射肿瘤学家的指导下进行。有关该领域的综述和治疗指南可参考(J. Dai and M. Duvic, “Cutaneous T-Cell Lymphoma: Current and Emerging Therapies,” Oncology (Williston Park) 37 (2023): 55–62.和208. J. Latzka, C. Assaf, M. Bagot, et al., “EORTC Consensus Recommendations for the Treatment of Mycosis Fungoides/Sezary Syndrome—Update 2023,” European Journal of Cancer 195 (2023): 113343)。
局部治疗
局限期MF的一线治疗为局部皮质类固醇。在一项非对照前瞻性研究中,85%的1A/B期患者使用卤倍他索丙酸酯,总反应率为94%,且几乎无毒性。另一种局部用药选择为0.02%的氮芥凝胶。在一项2期试验中,1A-IIA期MF患者每天使用0.02%凝胶治疗,最长达12个月。58.5%的患者观察到反应,其中13.8%达到完全反应;85.5%的患者观察到持续反应,最常见的不良反应是接触性皮炎和刺激性皮炎。对于难治性和持续性的皮肤病变,可以考虑使用1%贝沙罗汀局部凝胶。前瞻性试验已显示出44%至63%的总反应率。局部使用Toll样受体(TLR)激动剂可导致局部产生干扰素和其他细胞因子,诱导细胞死亡,并促进宿主抗肿瘤免疫,已在局限期MF中显示出疗效。例如,20名1A-2B期患者接受5%咪喹莫特(TLR7激动剂)治疗,观察到80%的总反应率,其中包括45%完全反应。毒性反应有限,包括局部疼痛、红斑、溃疡和瘙痒。虽然有报告称出现流感样症状和疲劳等全身性症状,但症状较为罕见。大多数不良事件为自限性,并在治疗后的最初几周内自行缓解。Resiquimod是一种强效TLR7/8激动剂,在一项1期试验中使用了0.03%和0.06%的局部Resiquimod凝胶。在12名接受治疗的患者中,75%的受累部位观察到临床改善,90%的患者在受累部位的恶性T细胞克隆数量减少,并且观察到了一种超靶区的、可能是免疫介导的效果。
光疗
光疗是一种重要的治疗手段,可以单独使用,也可以与局部治疗联合用于有限阶段疾病的患者,包括窄波紫外线B(NBUVB,311 nm)和8-甲氧基补骨脂素加紫外线A(PUVA)。NBUVB用于斑块和斑疹期MF。PUVA是有色皮肤的首选光疗方式。光疗在许多回顾性和前瞻性研究中显示出疗效,并且最近发表了一份关于光疗使用的全面共识声明。
放疗
MF/SS对放射线敏感,因此对于局限性、单发病变的MF患者,可以考虑以治愈为目的的放射治疗。对于病变更广泛的患者,姑息性局部放射治疗或低剂量全身皮肤电子束治疗(TSEBT)有效。
进展期MF/SS的治疗
概述
进展期MF/SS患者需要多学科协作,因为这些患者的管理通常需要多种皮肤定向治疗、生物反应调节剂,以及最终顺序使用系统药物(图1)。与局限期疾病一样,多药化疗并不合适;通常更倾向于采用与NCCN指南一致的“风险适应性”和基于分期的方法,包括生物反应调节剂(例如贝沙罗汀和干扰素α)、组蛋白去乙酰化酶抑制剂(例如romidepsin)或单克隆抗体或抗体-药物偶联物(例如莫格利珠单抗、维布妥昔单抗)。治疗决策需个体化,基于患者的年龄、体能状态、疾病负荷的程度、疾病进展的速度以及既往治疗。尽管全球的处方模式差异很大,但并未观察到生存差异。受累的组织部位(即皮肤、血液、淋巴结和/或内脏)以及最急需治疗的组织部位也是治疗决策中的重要因素,因为许多药物在不同组织部位的效果并不相同。例如,在肿瘤期但无血液、淋巴结或内脏受累患者中(II期B),维布妥昔单抗的总反应率为40%,而在更晚期(>II期B)疾病的患者中,反应率≤15%。相比之下,莫格利珠单抗在血液、皮肤和淋巴结/内脏隔室中的总反应率分别为68%、42%和≤17%。
贝沙罗汀
内源性视黄醇类化合物全反式视黄酸和9-顺式视黄酸(即维生素A衍生物)在与两种类固醇激素受体家族——视黄酸受体(RAR)和视黄醇X受体(RXR)结合后,调节从胚胎发育到细胞生长、分化和存活的广泛生物过程。这些受体形成同源或异源二聚体后,根据是否与配体结合,招募各种核共抑制或共激活蛋白。多种RAR视黄醇类化合物已用于MF/SS,无论是局部还是全身给药,反应率均超过50%。
然而,1999年,口服RXR选择性贝沙罗汀被FDA批准用于CTCL,并后来被批准为局部凝胶制剂。实验室研究表明,贝沙罗汀在CTCL细胞系中促进细胞周期阻滞和凋亡。在一项多中心II-III期研究中,94名接受过中位数五种既往治疗的晚期CTCL患者,其中绝大多数对至少一种既往系统治疗耐药,每天接受至少300 mg/m²的口服贝沙罗汀。在以300 mg/m²剂量治疗的患者中,观察到总反应率为45%,其中只有2%为完全反应。
尽管使用更高剂量时观察到总反应率有所提高,但差异并无统计学意义,而剂量限制性毒性在这些患者中更为常见(50%对89%)。尽管可能存在剂量-反应关系,但300 mg/m²剂量似乎提供最佳的风险-收益比。治疗相关的最常见毒性包括高甘油三酯血症(82%)和中枢性甲状腺功能减退(29%)。骨髓抑制很少见,通常不会引起严重问题。由于高甘油三酯血症引起的胰腺炎可能很少观察到,但在停止治疗后是逆,在开始治疗前应进行基线血脂检查和游离T4/TSH检查。在一项回顾性研究中,所有接受贝沙罗汀治疗的患者都出现高脂血症和中枢性甲状腺功能减退,通常在开始治疗后的几周内发生。因此,通常建议在开始贝沙罗汀治疗前使用降脂药物(例如非诺贝特)和低剂量左甲状腺素(例如50微克)。在临床实践中,贝沙罗汀通常以较低剂量150 mg/m²开始,并在治疗4周后根据患者耐受性逐渐增加到全剂量。大多数反应发生在治疗开始后的2-3个月内,但也可能会延迟。因此,在没有疾病进展或毒性的情况下,治疗应持续长达6个月。对于有反应的患者,应继续治疗直至疾病进展,并且根据反应的质量,应考虑联合皮肤定向治疗(例如NB-UVB,PUVA)。最近发表了关于贝沙罗汀实验室监测、支持治疗和安全临床处方的指南(J. J. Scarisbrick, S. Morris, R. Azurdia, et al., “U.K. Consensus Statement on Safe Clinical Prescribing of Bexarotene for Patients With Cutaneous T-Cell Lymphoma,” British Journal of Dermatology 168 (2013): 192–200)。
HDAC抑制剂
组蛋白去乙酰化酶(HDACs)催化从组蛋白和非组蛋白上去除乙酰基团。由于组蛋白乙酰化与开放的染色质构型相关,这种构型与活跃的基因转录有关,因此HDACs通过促进组蛋白去乙酰化和基因转录的表观遗传抑制发挥作用。由于HDACs调节多种与致癌过程相关的机制,HDAC抑制剂的临床活性可能通过多种机制实现,包括改变细胞周期和凋亡调节蛋白的基因表达、调节细胞生长和存活的非组蛋白的乙酰化、血管生成、聚集体形成以及DNA修复。此外,HDAC抑制剂对CTCL中的肿瘤微环境有深远影响。
Vorinostat和Romidepsin是两类I和II型HDAC抑制剂(即泛HDAC抑制剂),前者在多种淋巴瘤亚型中广泛表达。Vorinostat和罗米地辛的早期I期研究确立了它们在淋巴增殖性疾病(包括CTCL)中的安全性和潜在疗效,从而为更大规模的II期研究铺平了道路。一项早期II期研究确定了每天一次口服400毫克Vorinostat为最佳剂量,该剂量在74名既往接受过治疗的CTCL患者中进行了进一步研究,其中大多数(>80%)为进展期。晚期疾病患者的总反应率约为30%,并且中位反应持续时间估计超过185天。然而,值得注意的是,在MAVORIC研究中,使用更新的反应标准观察到的Vorinostat反应率显著较低(即<10%)。大多数反应迅速(即<2个月),并且在肿瘤期疾病和Sézary综合征患者中也观察到。未达到客观反应的患者似乎从治疗中获得了临床获益,包括疾病稳定、淋巴结肿大减轻和瘙痒缓解。最常见的非血液学不良事件(在近50%的患者中观察到)是胃肠道毒性(恶心、呕吐、腹泻)。血液学毒性,包括贫血或血小板减少,在高达20%的患者中观察到。在有反应的患者中,长期使用伏立诺他似乎耐受性良好。QT间期延长很少观察到,但对于有QT延长风险的患者,建议进行监测和适当的电解质补充。
Romidepsin通过4小时静脉输注(14 mg/m²)在每4周的第1、8和15天给药,在两项II期研究中进行了评估,其中最大的一项研究包括96名患者,大多为进展期;进展期患者的总反应率为38%,中位反应持续时间超过1年。观察到的毒性特征与伏立诺他描述的相似。对这些患者的一个子集进行的密集心脏监测未能发现任何具有临床意义的心脏毒性。在标准剂量诱导后,一部分MF/SS患者可能通过减量“维持”(每2周或4周)给药实现持久缓解。例如,在38名MF/SS患者中,17名实现了持久(>6个月)缓解,其中9名通过减量、剂量节省的方案维持。在实现持久缓解的患者中,治疗的中位持续时间为15个月(范围:7–34个月)。
其他HDAC抑制剂(包括强效泛HDAC抑制剂)也在CTCL中显示出活性。需要进一步研究以充分明确CTCL中对HDAC抑制剂耐药的机制,从而促进开发合理联合应用HDAC抑制剂的治疗方案。
干扰素
干扰素(例如干扰素α-2b、干扰素γ-1b)在CTCL中具有多种作用和免疫调节功能,ORR高达50%~70%,CR率高达20%~30%,尤其是在局限期患者中。尽管通常被认为是局限期CTCL的二线治疗,但干扰素α(通常每天剂量为300万至1000万单位,每周3次)在晚期患者的一线治疗中也值得考虑。反应可在几个月内实现,肿瘤期MF和SS患者可获得反应,且偶尔较为持久。此外,干扰素α可以成功地与其他常用于这些患者管理的治疗方式联合使用,包括PUVA、贝沙罗汀、化疗和体外光化学疗法(ECP)。例如,在一组51名大多为进展期的患者中,接受单药低剂量干扰素α治疗,34名(67%)有反应,其中21名(41%)CR,9名长期缓解。同样,在一组47名III/IV期患者中,89%有外周血受累,接受ECP联合干扰素α治疗的患者反应率超过80%。干扰素α与骨髓抑制、转氨酶升高以及高剂量时的剂量限制性流感样副作用相关。
体外光化学疗法
在体外光化学疗法(ECP)中,将汇集的白细胞分离和血浆分离产品暴露于8-甲氧基补骨脂素(8-MOP),然后通过体外循环,通过一个1毫米厚的可丢弃盒暴露于UVA辐射。随后将经过照射的白细胞(约占外周血白细胞的5%)重新输回体内。补骨脂素在UVA暴露后与DNA共价结合并交联,通过多种机制(涉及bcl-2家族成员、线粒体膜电位的破坏以及外源性细胞死亡途径)诱导大多数治疗淋巴细胞凋亡。相比之下,ECP可导致单核细胞激活,包括基因表达的显著变化,以及树突状细胞分化,这被认为最终会增强抗原呈递并启动宿主免疫反应。
在Edelson及其同事描述的37名红皮病型CTCL患者中有27人对ECP有反应的里程碑研究之后,ECP被美国食品药品监督管理局批准用于治疗CTCL,并且现在是许多中心治疗Sézary综合征的一线选择。此外,回顾性研究显示,与大多数系统治疗(包括HDAC抑制剂)相比,ECP在至下次治疗时间上具有优势。
尽管反应在病例系列之间有所不同,但ORR约为60%,CR率约为20%。由于当前的治疗方案不再需要口服8-MOP,从而消除了恶心,ECP安全且通常非常耐受。长期使用ECP可能会导致缺铁性贫血。尽管其作用机制尚未完全明确,但有证据表明ECP具有免疫调节作用,可能增强宿主的抗肿瘤免疫。因此,ECP治疗后至缓解的中位时间约为6个月。接受ECP治疗的患者的中位生存期超过8年,而在CR者中,许多人可持久缓解,从而可能使部分患者能够减少对CTCL定向治疗的依赖。
在一项回顾性研究中,早期接受ECP治疗(即在前三种治疗方案内)的患者与接受其他药物或在治疗过程中较晚接受ECP的患者相比,至下次治疗的中位时间更长(接近4年)。尽管预测治疗反应的患者或疾病特异性因素并不完美,但没有显著淋巴结或内脏受累的Sézary综合征患者如果在确诊后及时开始ECP治疗,可能更有可能产生反应。此外,没有严重免疫缺陷的患者(表现为正常或接近正常的细胞毒性T细胞和CD4/CD8值,且没有系统化疗的既往暴露),可能更有可能对治疗产生反应。虽然ECP作为单药治疗有效,但它也已与其他治疗策略联合使用,包括干扰素、贝沙罗汀和TSEBT。
莫格利珠单抗(Mogamulizumab)
莫格利珠单抗(KW-0761)是一种针对趋化因子受体CCR4的人源化单克隆抗体,经过脱岩藻糖处理,因此具有增强的抗体依赖性细胞介导的细胞毒性(ADCC)。在一项I/II期研究中,莫格利珠单抗耐受性良好,ORR为37%。在日本一项II期研究中,ORR为29%(2/7),均为PR。除了通过ADCC清除恶性T细胞外,莫格利珠单抗还可能抑制调节性T细胞(Treg)介导的免疫抑制,并且可能需要与免疫调节疗法(包括免疫检查点抑制)进一步研究。一项随机的III期临床试验(MAVORIC)比较了莫格利珠单抗与伏立诺他治疗复发/难治性CTCL的效果,结果显示莫格利珠单抗组的MF/SS患者无进展生存期显著改善。接受莫格利珠单抗治疗的患者在血液中的ORR(68%)高于皮肤(42%)或淋巴结(17%)的缓解率。因此,SS患者的ORR最高(37%)。总体而言,莫格利珠单抗治疗耐受性良好,很少出现≥3级不良事件(AE)。输液相关反应是最常见的1级或2级AE,发生率为32%。莫格利珠单抗可发生皮疹,并且在临床上和组织病理学上可能类似于CTCL,但可以在不停止治疗的情况下进行管理。治疗相关的皮疹以巨噬细胞和CD8+ T细胞浸润为主,并且与Sézary综合征患者中更好的疾病控制相关。这些积极的结果促使莫格利珠单抗在2018年获得FDA批准,用于至少接受过一次系统治疗失败的MF/SS患者。在SS中,对于达到缓解的患者,在治疗停止后可以实现持久缓解(>12个月),并且在随后复发时可以成功重新开始治疗。
Alemtuzumab
Alemtuzumab是一种针对CD52的人源化IgG1单克隆抗体,CD52是一种在B细胞、T细胞和单核细胞上广泛表达的抗原。在一项针对22名晚期MF/SS患者的II期研究中,ORR和CR率分别为55%和32%,中位至治疗失败时间为1年。鉴于存在感染并发症的显著风险,研究了14名SS患者(其中大多数为复发/难治性疾病患者)接受低剂量皮下注射Alemtuzumab,大多数患者在第1天接受3 mg皮下Alemtuzumab注射,随后每隔一天接受10 mg剂量,直至Sézary细胞计数<1000/mm³。除了1例患者的最佳反应为稳定外,10名患者中有9名获得了缓解,其中3名为CR。对于大多数患者,至治疗失败时间超过12个月。然而在接受最低剂量(即10 mg)Alemtuzumab治疗的患者中未观察到感染并发症。
在一项接受改良低剂量皮下Alemtuzumab治疗6周的小型患者队列中,也报告了类似的结果,且未观察到感染并发症。除了血液学毒性外,晚期MF/SS患者接受常规剂量Alemtuzumab治疗还与高感染并发症发生率相关。总体而言,接受治疗的患者中有三分之二发生感染并发症,其中大多数为细菌感染,包括败血症。巨细胞病毒(CMV)再激活是最常见的病毒感染。此外,还观察到了肺孢子菌肺炎和侵袭性真菌感染。因此,对于接受Alemtuzumab治疗的患者,应常规给予复方磺胺甲噁唑用于肺孢子菌肺炎(PJP)预防,以及阿昔洛韦用于单纯疱疹病毒/水痘-带状疱疹病毒(HSV/VZV)预防。此外,应每1–2周通过定量PCR进行CMV监测,并在病毒再激活时启动更昔洛韦或口服缬更昔洛韦的抑制治疗。对于接受适当支持治疗的晚期MF/SS患者,低剂量皮下Alemtuzumab似乎安全有效。
维布妥昔单抗
鉴于在II期研究中观察到的维布妥昔单抗(BV)的有希望的缓解率,一项随机的III期临床试验(ALCANZA)将BV与研究者选择(甲氨蝶呤或贝沙罗汀)进行比较,结果显示BV组患者的PFS显著改善(>12个月对比3.5个月),并促成了其在既往接受过治疗的CTCL患者中获批。在用BV治疗的局限期MF患者中,40%的患者观察到持续至少4个月的反应(ORR4),而在肿瘤期(II期)患者中,ORR4为63%。与“高CD30”淋巴瘤的先前经验一致,原发性皮肤ALCL患者(疾病仅限于皮肤)的ORR4为89%。
最近,在中位随访时间为46个月的报道中,最终的ALCANZA数据确认了BV的获益。在MF患者中,接受BV治疗的患者的中位PFS为16.1个月,而接受甲氨蝶呤或贝沙罗汀治疗的患者为3.5个月。因此,治疗后1年,34.5%的BV患者需要使用其他系统治疗药物,而86.6%的甲氨蝶呤或贝沙罗汀治疗患者需要其他治疗方法。与CD30表达或大细胞转化的存在无关,均观察到了BV的获益。标准免疫组化可能低估了CD30表达的程度,并且预计患者体内CD30表达随时间和不同疾病部位存在显著的变异性。此外,CD30表达是动态的,在CD30缺失、“低”(<5%–10%)和“高”CD30表达的患者中均观察到反应。因此,对于符合GV治疗条件的患者,治疗决策并不依赖于CD30状态。
Denileukin Diftitox-Cxdl(E7777)
CTCL克隆T细胞表达三聚体IL-2受体复合物的成分,包括用于高亲和力结合的α链(CD25)、β链(CD122)和共同γ链(CD132)。使用单克隆抗体Daclizumab靶向IL-2受体在成人T细胞白血病/淋巴瘤中安全且部分有效。为了改善这些针对IL-2受体的结果,开发了denileukin diftitox(Dd)。Denileukin diftitox是一种融合蛋白,由人类IL-2与截短的白喉毒素融合而成,对IL-2受体具有高亲和力,并在受体结合后内化,从而释放毒素并诱导凋亡。I/II期研究显示约三分之一的患者ORR,随后进行的III期研究将71名CD25阳性(即免疫组化显示≥20% T细胞阳性)的CTCL患者(大多数为晚期疾病)随机分配到每天9或18 μg/kg的Dd,通过静脉注射,每3周连续给药5天,最多8个周期。30%的患者获得ORR(20% PR,10% CR),另外32%的患者病情稳定。中位至缓解时间为6周,95%的反应患者在第9周时显示出反应迹象。中位反应持续时间为7个月(范围2.7–46.1个月)。在反应者中,观察到生活质量的显著改善。一项大规模的III期安慰剂对照试验中,ORR为44%,中位PFS超过2年。尽管这些研究仅纳入CD25阳性患者,但从不同部位或不同时间获取的活检显示显著的变异性,表明CD25阴性CTCL患者也可能从治疗中受益。
在一项前瞻性分析中,CD25阳性和阴性淋巴瘤之间的ORR存在显著差异,仅有20%的CD25缺失或低表达患者对治疗有反应,而CD25阳性疾病的患者反应率接近80%。在一项包括307名患者的三项试验的荟萃分析中,接受Dd治疗的CD25阳性患者的ORR为47.5%,中位PFS超过2年。相比之下,CD25阴性患者ORR仅30.6%,PFS超过487天。对于接受安慰剂治疗的患者(n=44),报告的反应率为15.9%,中位PFS为4个月。值得注意的是,在初次反应后复发的患者中,再次接受Dd治疗时也观察到反应。
大约25%的患者在治疗的前几个周期内出现血管渗漏综合征,定义为出现三种症状,包括低血压、水肿和低白蛋白血症(白蛋白<3 g/dL)。大多数这些患者在后续治疗中未出现进一步症状。大约三分之二的患者在治疗后1小时内出现输液反应,但皮质类固醇预处理可以缓解。最近,一种纯度更高的denileukin diftitox-Cxdl(E7777)已完成临床试验,在III期研究中取得了与历史数据相当的疗效结果,并于2024年获得FDA批准用于至少接受过一次先前治疗的I至III期CTCL患者。
免疫检查点抑制剂
通过免疫调节疗法(包括ECP和干扰素-α)可以实现持久缓解。尽管报道极少,但它们表明,当适当调动宿主免疫时,可以在特定患者中实现持久反应。这些结果加上相当一部分患者中高水平的PD-L1表达以及另一些患者中的PD-L1结构变异,为检查点阻断(CPB)提供了有力的理由。尽管在早期临床试验中纳入的CTCL患者较少,但已观察到持久反应(表2)。例如,在一项针对重度经治患者的II期研究中,接受帕博利珠单抗治疗的晚期患者的ORR为38%。具有大细胞转化(LCT)的MF在遗传上较为复杂(具有高突变负荷),经常下调MHC I类分子,并且表达PD-L1,包括PD-L1结构变异,所有这些都与免疫逃逸一致。尽管在LCT患者中的经验有限,但现有证据支持在这些患者中使用检查点阻断的合理性。在CTCL中,“超进展”的风险较低,正如在许多实体瘤中一样,而目前在CTCL中的证据仅限于个案报告。
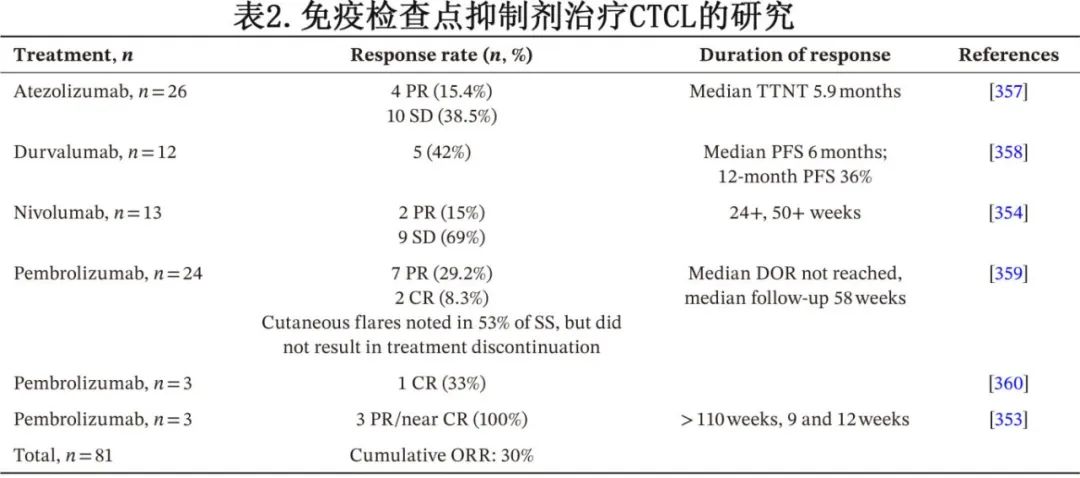
相比之下,“肿瘤炎症”现象较为常见,可导致皮肤触痛和瘙痒加重,可能难以与疾病进展区分开来,尤其是在恶性T细胞表达PD-1的患者中。不应因炎症反应而停止治疗,因为这些反应可以通过支持治疗来管理,包括使用外用皮质类固醇。这些令人鼓舞的结果,加上目前可用的多种免疫调节剂,为未来的联合策略提供了基础,并且目前正在进行相关研究。
全身化疗
CTCL对传统化疗药物的反应很少持久,其中位时间至下次治疗仅以月为单位,超过90%的患者在治疗的第一年内需要额外治疗。此外,将系统化疗作为一线治疗与死亡率增加相关。因此,多药化疗很少使用,更倾向于使用新的治疗药物,包括参与临床试验。由于对于需要系统化疗的MF/SS患者没有标准治疗方案,并且开始治疗的决定是个性化的,包括考虑与先前治疗相关的反应和并发症,因此应参与设计良好的临床试验。
普拉曲沙是一种新型抗叶酸药物,对还原型叶酸载体(RFC-1)具有高亲和力,并且与甲氨蝶呤相比具有新的耐药机制,在PROPEL研究中ORR为29%。该研究主要由外周T细胞淋巴瘤患者组成,其中大多数患有难治性疾病。值得注意的是,研究中纳入12名患有转化型MF的患者。这些患者中的许多人都接受过超过五种先前的系统治疗,包括CHOP或类似CHOP的方案。除了一例患者外,这些患者对其最近的治疗均耐药。根据研究调查员的评估,58%的患者缓解,中位缓解持续时间和PFS为4–5个月。
一项剂量探索研究的结果在更大规模的CTCL患者队列中报告,确定的最佳剂量为15 mg/m²,每周给药3次,每4周为一个周期,ORR为43%。除了补充叶酸和维生素B12外,口服亚叶酸(25 mg,每天3次,第2天和第3天)也可显著降低黏膜炎的发生率。
其他治疗方法,包括蛋白酶体抑制、免疫调节策略以及更具针对性的方法,值得进一步研究。单药吉西他滨(1200 mg/m²,在28天周期的第1天、第8天和第15天给药)通常耐受性良好,ORR约为70%,其中大多数为PR,中位持续时间约为1年。聚乙二醇化脂质体阿霉素(每2周给药一次,剂量为20 mg/m²)与普拉曲沙相当的ORR(约为40%)相关,其中很少有CR。最近的真实世界经验表明,在皮肤部位的反应率更高,包括在肿瘤期疾病患者中高达76%的反应率,但在其他部位的反应率则显著较低(<30%)。发生过死亡案例被归因于心脏毒性(罕见)。
异基因造血干细胞移植
关于大剂量化疗和自体干细胞移植的经验大多局限于病例报告,表明治疗后的缓解通常短暂。相比之下,异基因移植后观察到的持久缓解可通过移植物抗淋巴瘤免疫反应来解释。最近报告了一项对60名晚期MF/SS患者进行异基因干细胞移植的回顾性分析。在该研究中,患者在移植前中位接受过4种既往治疗,随后接受减低强度预处理(73%)或清髓性预处理(27%),并进行了亲缘供者(75%)或相合无关供者(25%)移植。在接受减低强度预处理或HLA相相合/亲缘供者干细胞的患者中,1年非复发死亡率为14%,而在接受清髓性预处理或相合无关供者移植的患者中为38%–40%。在疾病早期阶段(定义为首次或第二次缓解或在3种或更少系统治疗后复发)进行移植与较低的复发率相关(1年时25%对比44%),并且3年OS有统计学上不显著的提高(68%对比46%)。鉴于非复发死亡率的差异,减低强度预处理和使用相合亲缘供者均与更好的OS相关(3年OS率为63%)。在26名复发的患者中,有17名接受了供体淋巴细胞输注,其中47%实现CR,从而为MF/SS中的移植物抗淋巴瘤效应提供了证据。估计的3年PFS率和OS率分别为34%和53%。最近对EBMT经验的更新再次表明,异基因干细胞移植在少数患者中可以治愈,但非复发死亡率和疾病进展仍是挑战。在CIBMTR的一个大型系列研究中(n=129)观察到类似结果,1年NRM和疾病进展率分别为19%和50%,5年PFS和OS分别为17%和32%。一项系统综述和荟萃分析汇总了来自五项研究(266名患者)的数据,显示异基因移植后的复发率为47%,NRM为19%。鉴于有可能实现完全且持久的缓解,异基因干细胞移植可以在特定患者中考虑,并且最近已发布了帮助患者选择的共识指南,但应在有经验的学术中心进行。
临床试验
生活质量(QOL)和支持治疗是治疗选择和CTCL管理中至关重要的因素。因此,患者受益于多学科协作,由经验丰富的皮肤病理学家、皮肤科医生以及内科和放射肿瘤学家提供意见。治疗通常是逐步升级和连续进行的,并且在适当选择的患者中,有时会重复使用。尽管目前可用药物的大多数反应并不完全或持久,但在局限期患者中,病情稳定可能与生活质量改善相关。对疾病发病机制的深入理解,包括对CTCL中遗传背景和肿瘤微环境作用的认识,已经确定了新的治疗机会,其中许多是正在进行的临床试验的主题。因此,临床试验参与将在CTCL患者的多学科管理中继续发挥越来越重要的作用,并且强烈鼓励患者参与。
参考文献
Hristov, A.C., Tejasvi, T. and Wilcox, R.A. (2025), Mycosis Fungoides, Sézary Syndrome, and Cutaneous B-Cell Lymphomas: 2025 Update on Diagnosis, Risk-Stratification, and Management. Am J Hematol. https://doi.org/10.1002/ajh.27735

 分享
分享
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#原发性皮肤淋巴瘤#
4 举报