
肝豆状核变性:研究新进展与诊疗误区
2024-09-01 神经科学论坛 神经科学论坛 发表于上海

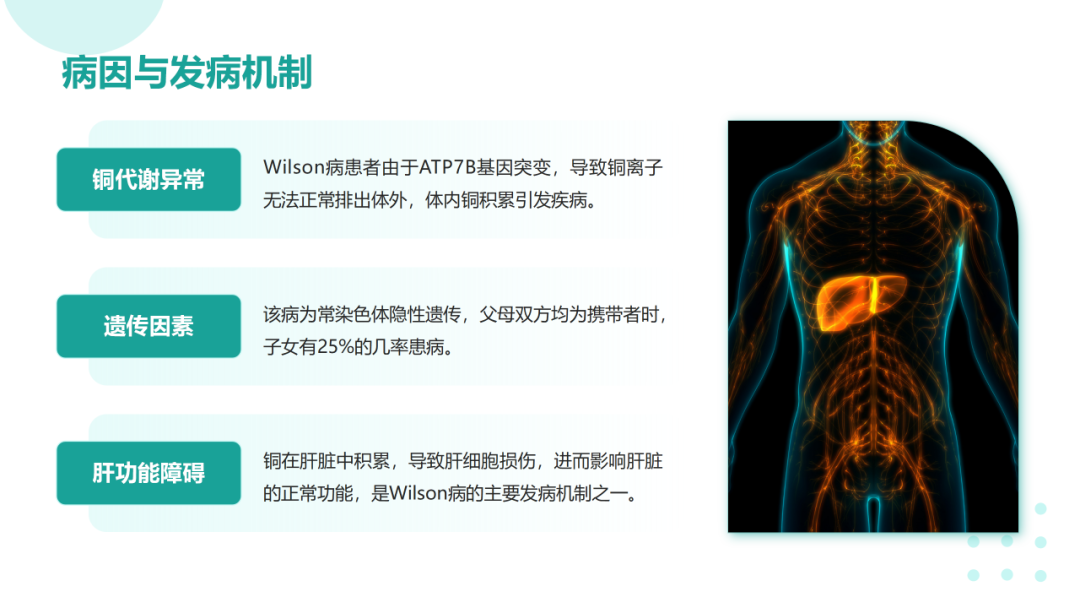
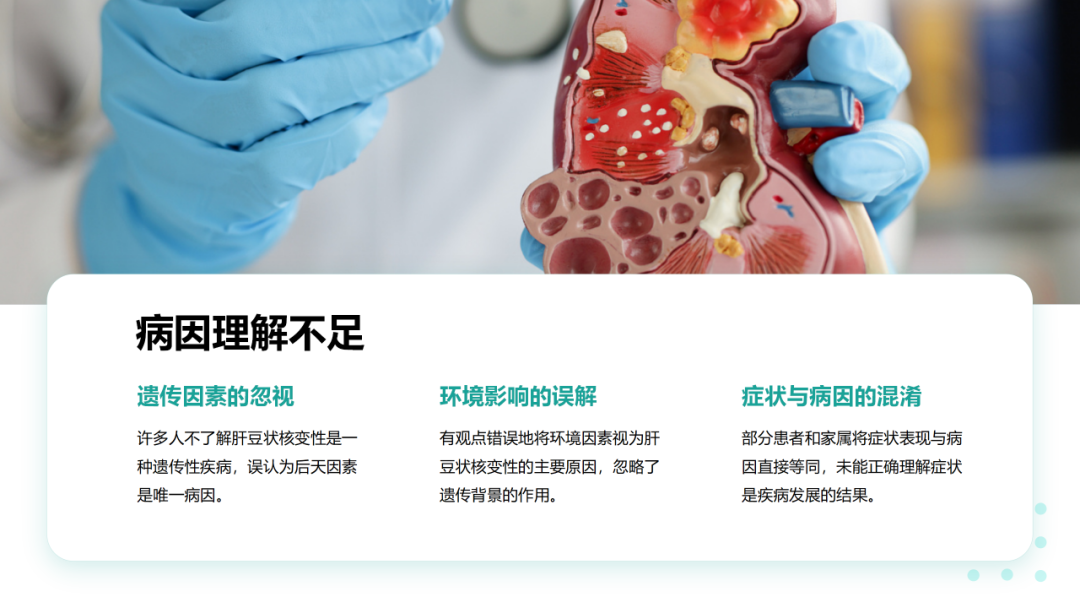
肝豆状核变性是一种常染色体隐性遗传的铜代谢障碍疾病。致病基因为ATP7B,其突变导致ATP酶功能减弱或丧失,使血清铜蓝蛋白合成减少以及胆道排铜障碍,导致蓄积于体内的铜离子在肝、脑、肾、角膜等处沉积。
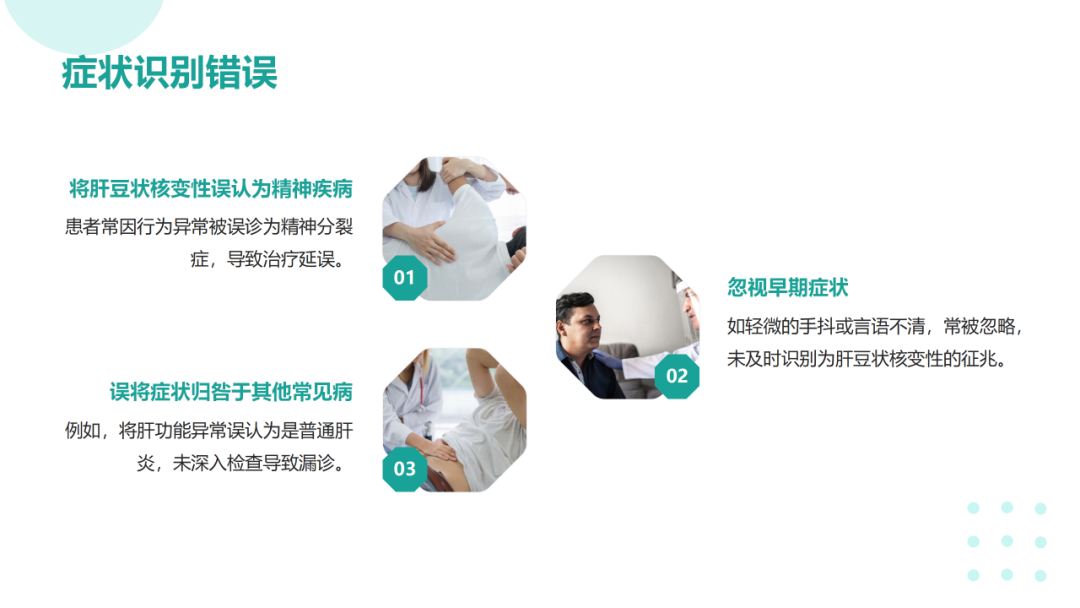
论坛导读:肝豆状核变性是一种常染色体隐性遗传的铜代谢障碍疾病。致病基因为ATP7B,其突变导致ATP酶功能减弱或丧失,使血清铜蓝蛋白合成减少以及胆道排铜障碍,导致蓄积于体内的铜离子在肝、脑、肾、角膜等处沉积。WD患者临床表现多样,因受累器官和程度不同而异,主要表现为肝脏和/或神经系统损害表现,此外,还会伴有其他系统损害现象。包括神经精神症状、肝生化异常、角膜K-F环、肾损害、溶血性贫血、骨骼肌肉损害等表现。临床上分为肝型、脑型、其他类型及混合型。
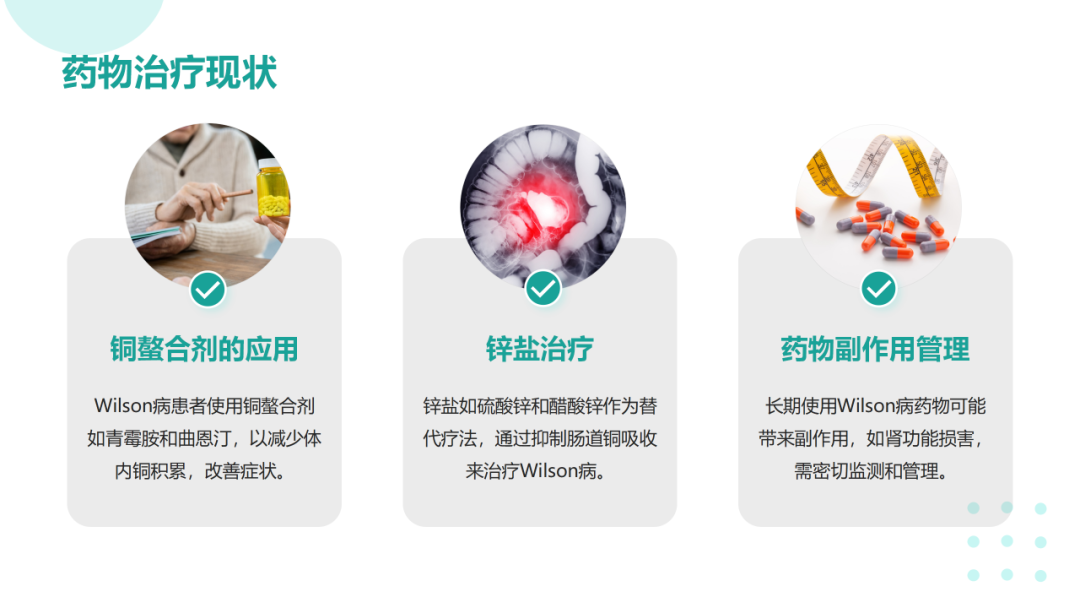
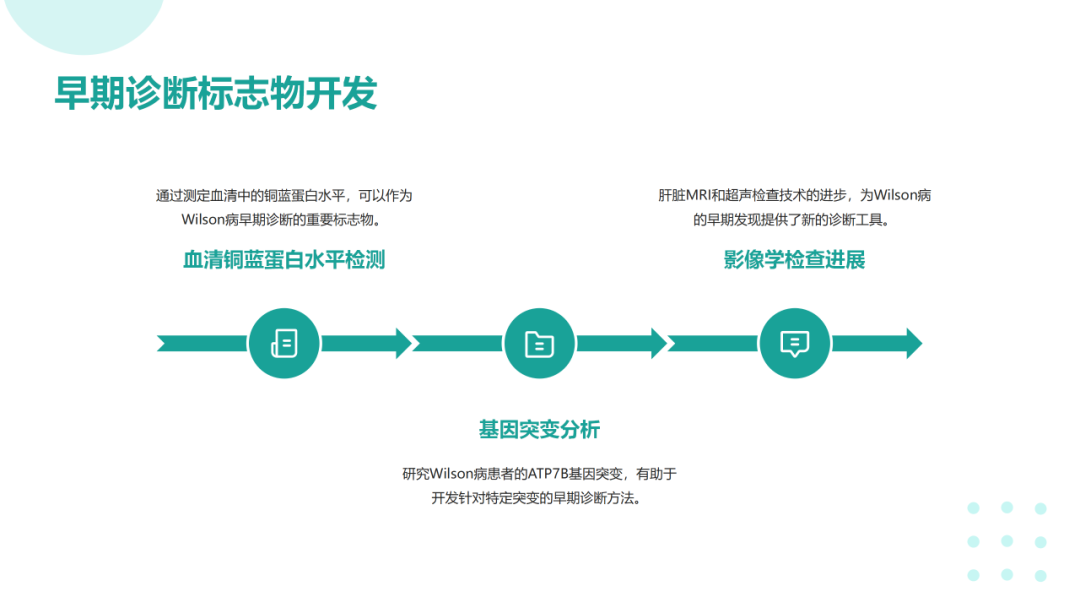
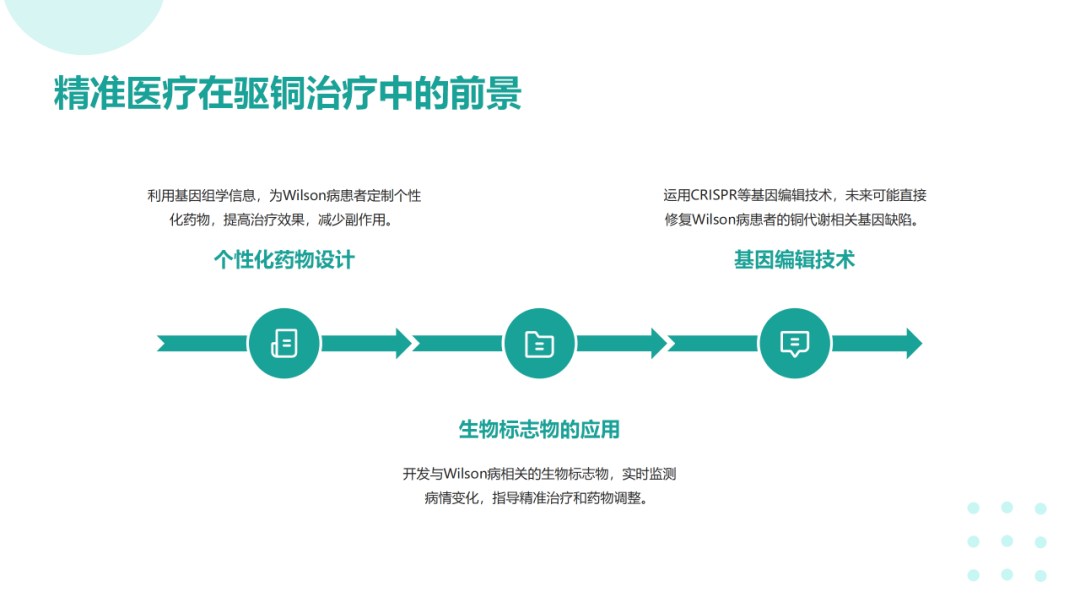
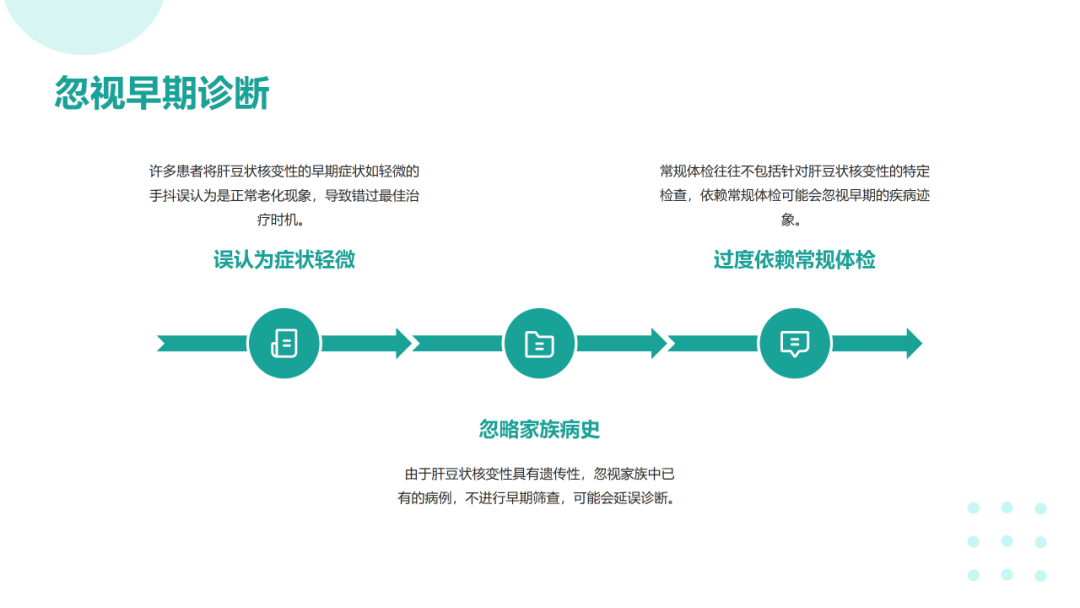
肝豆状核变性可在任何年龄发病,多发于年龄<40岁的患者。该病可累及全身多个脏器,临床体征和症状差异较大,最常表现为肝脏(包括肝硬化)和神经系统受累;眼部征象(Kayser-Fleischer环);溶血伴急性肝衰竭。肝豆状核变性的临床症状可不典型,因此对肝功能异常不能用常见肝病解释的患者应及时行K-F、血清铜蓝蛋白、24h尿铜、腹部超声、头颅MRI等检查协助诊断。肝豆状核变性的治疗目的是减少铜摄入,阻止铜吸收,排出体内多余的铜,维持体内铜代谢平衡。一经诊断,应及早治疗,在医生指导下终身低铜饮食和药物治疗。症状前个体一般指以下3种情况:常规体检发现转氨酶轻度增高但无症状且行ATP7B基因筛查确诊;意外发现角膜K‐F环但无症状且行ATP7B基因筛查确诊;Wilson病先证者的无症状同胞行ATP7B基因筛查确诊。在神经系统症状出现前,头部CT可见一些病变,包括脑室扩大,MRI更为敏感。第三脑室扩张,丘脑、豆状核、苍白球可见局部病变,同临床亚型相关。

















































本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言












#肝豆状核变性# #ATP7B#
91